Mechanochemistry has emerged as a transformative frontier in chemistry, leveraging mechanical forces to induce chemical changes in molecules. Among the promising innovations in this domain is the ability to deliberately activate carbon-carbon (C–C) bonds—a type of bond traditionally viewed as robust and unyielding. Researchers at the University of Illinois Urbana-Champaign, alongside collaborators from MIT and Duke University, have crafted a compelling new approach that challenges conventional assumptions and simplifies the prediction of C–C bond reactivity under mechanical stress.
The innovation centers on a mechanophore named NEO, a molecular structure capable of releasing controlled amounts of carbon monoxide when mechanical force breaks specific C–C bonds. Such functionality opens pathways to revolutionary applications, particularly in targeted drug delivery within the human body, where the controlled release of therapeutic gases like carbon monoxide could treat various ailments efficiently and safely.
The Challenge of Predicting Mechanochemical Behavior
Despite the excitement around mechanophores like NEO, a major obstacle has hampered progress: predicting how and when mechanophores respond to mechanical forces. The vectors of force—whether pushing, pulling, or twisting—create highly unpredictable effects at the molecular level, demanding resource-intensive experimentation and complex computational calculations to understand their mechanochemical responses.
Traditional computational methods, while accurate, often produce data that are difficult to interpret intuitively. Without accessible models that distill these complexities into digestible insights, progress in designing new mechanophores remains slow, overly dependent on trial and error.
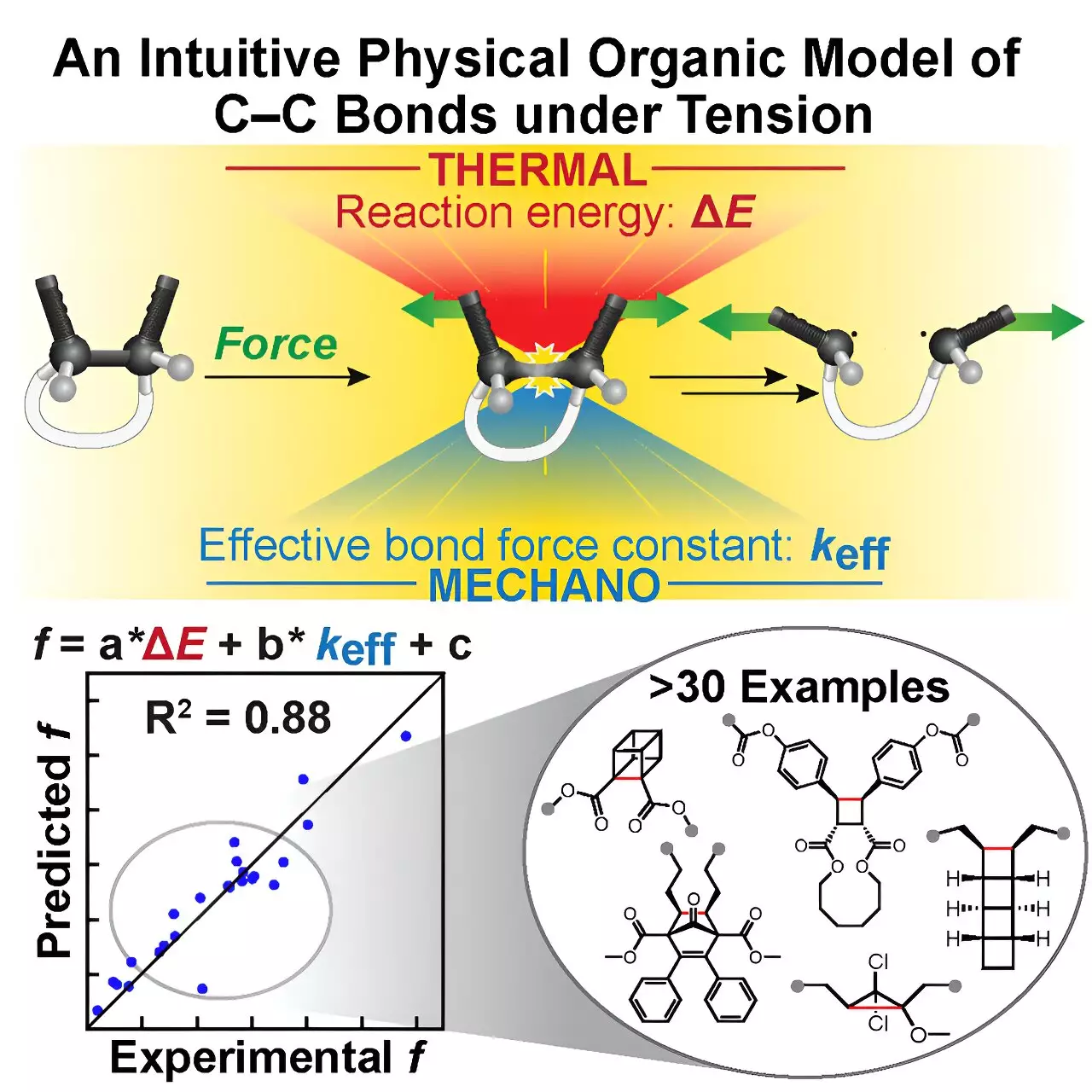
Introducing the Tension Model of Bond Activation (TMBA)
Against this backdrop, the team led by Prof. Jeffrey Moore, graduate student Yunyan Sun, and researchers Ilia Kevlishvili and Stephen L. Craig has presented the Tension Model of Bond Activation (TMBA)—a remarkably simple yet quantitatively robust tool for anticipating C–C bond behavior under tension.
Rooted in the classical Morse Potential, a model many chemistry students meet early in their education, TMBA offers an elegant analogy: the restoring force triangle. This conceptual framework breaks down complex molecular interactions into two core, easily computable parameters—the effective force constant and the reaction energy—that govern bond activation kinetics.
This approach transforms intricate computational outputs into a clear, mnemonic model, empowering chemists to grasp and anticipate mechanochemical reactions without being mired in dense data or specialized software. Yunyan Sun highlights this democratizing aspect: TMBA is not merely a high-level computational shortcut but an intuitive aid that bridges theory and practice, accelerating mechanophore design.
Simplification Without Sacrificing Precision
One might worry that simplifying mechanochemical kinetics into such a model could compromise accuracy. However, extensive validation tests have shown that TMBA’s predictions align quantitatively with experimental observations across more than 30 mechanophores, including four variants of NEO.
This balance between simplicity and precision marks a paradigm shift in the field. Much like how Lewis structures or Hammond’s postulate provide foundational understandings for chemical bonding and kinetics, TMBA offers a modular, teachable insight into the effects of mechanical forces on molecular bonds.
Co-author Ilia Kevlishvili emphasizes the significance of this conceptual clarity. Modeling mechanochemistry with such accessible tools can hasten discovery of novel materials with tailored responsiveness—heralding a new era of “productive mechanochemistry” where mechanical forces are harnessed deliberately rather than encountered as obstacles.
Collaborative Synergy Fuels Innovation
The trajectory of TMBA underscores the indispensable role of collaboration. The confluence of expertise from UIUC, MIT, and Duke’s MONET Center for the Chemistry of Molecularly Optimized NETworks created a fertile environment where experimentalists, theorists, and computational chemists converged.
Stephen L. Craig’s work on single-molecule mechanistic studies at Duke provided critical experimental validation, while Heather Kulik’s group at MIT contributed computational insights. The project’s success reflects not only the merging of technical skillsets but also a shared vision to reimagine C–C bond reactivity.
Such interdisciplinary cooperation exemplifies how complex scientific puzzles—like translating mechanical inputs into predictable chemical reactions—can only be unraveled when diverse perspectives meld into cohesive frameworks. This collective effort also ensures that the model is versatile across different molecular systems and adaptable for emerging challenges.
Changing the Paradigm: C–C Bonds Are Not Invincible
Perhaps the most striking implication of this research is an epistemological one: the long-held belief that C–C bonds are invulnerable to mechanical disruption must be revised. TMBA not only quantifies when and how these bonds can break but also provides a practical roadmap to exploit this susceptibility in designing new materials and therapeutic agents.
This insight carries profound ramifications across multiple sectors. In pharmaceuticals, the ability to trigger carbon monoxide release mechanically could revolutionize drug delivery, reducing systemic side effects and enhancing precision. In materials science, mechanophores designed with TMBA could yield self-healing polymers or stress-responsive coatings that extend product lifetimes.
A Teaching Moment for Future Chemists
Looking forward, Prof. Moore envisions TMBA as more than a research tool—it has the potential to become didactic gold. Given its grounding in fundamental chemical principles and clarity of application, TMBA could become a staple concept introduced even at the freshman college level, nurturing a new generation of chemists who intuitively understand the interplay between force and chemical reactivity.
This shift towards intuitive, accessible science is critical for accelerating innovation. When complex phenomena are distilled into memorable frameworks, ideas move faster from lab benches to real-world solutions.
By empowering chemists both practical and theoretical with fresh insights into the mechanical activation of bonds, TMBA stands to catalyze a fresh wave of discovery. The once immutable carbon-carbon bond now reveals itself as a gateway to dynamic, responsive chemistry.