Atoms, the fundamental building blocks of matter, are intricately complex. Each atom features a nucleus packed with positively charged protons and neutral neutrons, surrounded by a cloud of negatively charged electrons. The interaction between these electrons becomes even more intricate when atoms unite to form molecules. Consequently, simulating the behavior of molecules has emerged as one of the most challenging problems in modern science. This complexity doesn’t just stem from the intrinsic properties of atomic structures but also from the delicate interplay of quantum mechanics, making accurately predicting molecular behavior via traditional computational methods a daunting task.
Advancements through Machine Learning
In an exciting development, researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) and Google DeepMind have unveiled a cutting-edge machine learning algorithm capable of performing long-term molecular dynamics simulations with remarkable accuracy. Their findings, released in Nature Communications, mark a significant leap forward in molecular simulation techniques, holding implications for vital areas such as drug development and the optimization of materials including solar panels and batteries.
Traditional simulation techniques often revolve around solving the Schrödinger equation — a mathematical representation that defines the energy levels possible within quantum systems. This equation’s complexity means that even powerful computers struggle to provide solutions for molecules containing more than a handful of atoms, a task that can consume days or even weeks. As researchers endeavor to simulate longer time scales, the computational costs escalate, posing a major barrier for scientists hoping to understand phenomena like protein folding or molecular binding.
Revolutionizing Computational Chemistry
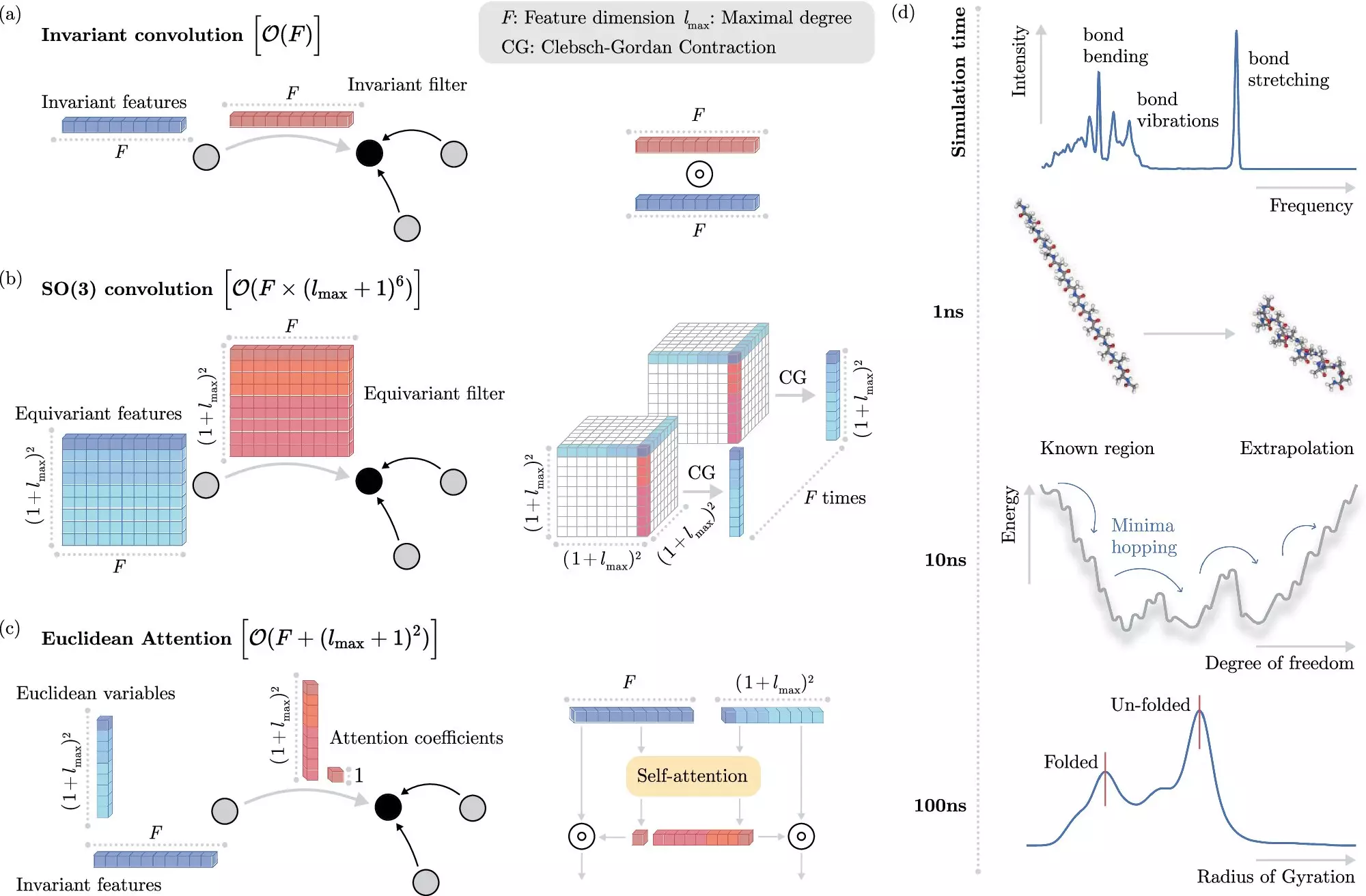
The genius of the recent innovation lies in the way it harnesses machine learning. Instead of directly grappling with the formidable Schrödinger equation, the newly developed algorithm learns to predict the outcomes of electronic interactions at an atomic level, significantly cutting down computation time and resources. This methodological shift highlights a key advancement in computational chemistry: the ability of machine learning models to forecast results without explicit modeling of electron interactions.
The real challenge, however, is teaching these machine learning systems to decipher the intricate dance between electrons without getting bogged down by computationally intensive processes. Here, the BIFOLD team’s innovative approach stands out. By decoupling spatial invariances — properties that remain constant under spatial transformations — from other data about chemical systems at the outset, they have streamlined the learning process. This advancement allows the model to apply its computational power to the most critical aspects of molecular interactions, vastly improving overall efficiency.
Groundbreaking Efficiency Gains
What once took months or even years of computation on high-performance clusters can now be executed within days, even on a standard computer node. This groundbreaking increase in efficiency heralds a new era for molecular dynamics simulations, enabling researchers to dive deeper into understanding the structure and dynamics of atomistic systems than ever before. “This newfound capability unlocks the potential for more comprehensive insights into fundamental natural processes,” remarks BIFOLD researcher Dr. Stefan Chmiela.
The implications of such advancements resonate strongly within pharmaceutical research. The precise simulation of molecular interactions within the human body could revolutionize drug development, eliminating the need for exhaustive experimental trials and yielding from a fiscal and environmental perspective.
Practical Applications and Future Directions
To illustrate the practical implications of this new algorithm, the research team applied their machine learning technique to identify the most stable variant of docosahexaenoic acid, an essential fatty acid associated with brain structure. The potential to analyze tens of thousands of options—something previously unfeasible using traditional quantum mechanics—demonstrates the expansive reach of this technology.
Prof. Dr. Klaus-Robert Müller, one of the leading scientists at Google DeepMind, succinctly captures the significance of the research: “This work embodies the transformation achievable when advanced machine learning aligns with physical principles, tackling long-standing challenges within computational chemistry.” The development sets the stage for the next generation of algorithms capable of accurately simulating larger molecular systems, particularly around complex long-range interactions.
The horizon looks bright as machine learning continues to penetrate the realms of quantum systems and molecular interaction. With each progressive step, researchers are not only striving to solve enigmatic challenges in chemistry but also improving efficiency and paving the way for innovations that could redefine our approach to science and technology.